
Sclerosi Laterale Amiotrofica (SLA)
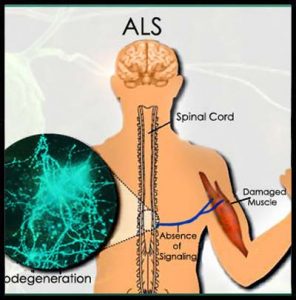
La Sclerosi Laterale Amiotrofica (SLA) è una malattia neurodegenerativa, ad andamento progressivo, caratterizzata dalla degenerazione primitiva dei motoneuroni superiori (centrali, siti a livello della corteccia cerebrale) e inferiori (periferici, siti nel tronco encefalico e nelle corna anteriori del midollo spinale), ovvero delle cellule nervose cerebrali e del midollo spinale specializzate nel controllo del movimento muscolare.
Clinicamente la malattia si esprime con disturbi piramidali agli arti, atrofia muscolare, soprattutto alle mani, fascicolazioni e deficit dei nervi cranici.
Contenuti dell'articolo
SLA – Epidemiologia
La SLA è una malattia rara con un’incidenza di 2-3 casi ogni 100.000 abitanti. La prevalenza nella popolazione generale è di 6-8 casi ogni 100.000 abitanti. L’associazione italiana sclerosi laterale amiotrofica ha stimato che la malattia colpisca circa 420 mila persone nel mondo, di cui 6000 solo in Italia (dati aggiornati al 2016). Ci sarebbero, inoltre 14 mila nuovi casi all’anno. E’ riportata un’alta incidenza nella Nuova Guinea Occidentale, nella penisola di Kii in Giappone e nell’isola di Guam: non se ne conosce il motivo.
L’età media di insorgenza è tra i 40 e i 70 anni; è più frequente negli uomini rispetto alle donne (con un rapporto M/F di 1.3). Le forme familiari costituiscono circa il 5% dei casi. E’ rarissima oltre gli 80 anni.
Sclerosi Laterale Amiotrofica – Cause
A oggi la causa della SLA è sconosciuta. Sono state formulate diverse ipotesi per spiegare la degenerazione motoneuronale che caratterizza la malattia: appare verosimile che molteplici meccanismi siano coinvolti e variamente embricati tra di loro.
L’ipotesi del danno ossidativo
In circa il 2% delle forme di sclerosi laterale amiotrofica sporadica e nel 15-20% delle forme familiari è stata riscontrata la presenza di una mutazione di un gene, localizzato sul cromosoma 21, che codifica per l’enzima superossido dismutasi, uno dei maggiori agenti antiossidanti cellulari. E’ verosimile, pertanto, che un malfunzionamento dei meccanismi anti-ossidativi esponga le cellule neuronali al danno tossico dei radicali liberi.
L’ipotesi del danno eccitotossico
Esistono evidenze sperimentali che dimostrano che il glutammato, insieme ad altri neurotrasmettitori a funzione eccitatoria, sono in grado di indurre la morte neuronale. In particolare, nei pazienti con SLA, esisterebbe un’alterazione del metabolismo del glutammato per cui il tuo trasporto nel sistema nervoso centrale sarebbe ridotto mentre la sua concentrazione nel liquor e nel sangue sarebbe aumentata.
L’ipotesi autoimmune
Una probabile correlazione con meccanismi di tipo autoimmune spiegherebbe la presenza di un’aumentata incidenza, nei malati di SLA, di linfomi Hodgkin e non Hodgkin (dal 10% al 30%).
L’ipotesi dei neurofilamenti
I neuroni più colpiti nel paziente affetto da sclerosi laterale amiotrofica sono i neuroni di maggior calibro che sono particolarmente ricchi di neurofilamenti: è stato ipotizzato che l’alterazione e il conseguente accumulo di neurofilamenti all’interno dei motoneuroni possa costituire un fattore nella genesi della malattia.
Ipotesi sui fattori ambientali
E’ stata ipotizzata la possibile correlazione tra l’esposizione ad agenti tossici quali alcuni fitofarmaci, in particolare insetticidi organoclorurati, e l’insorgenza di SLA. Tuttavia il ruolo dei fattori ambientali non è chiaro.
Sclerosi Laterale Amiotrofica – Aspetti Neuropatologici
I due elementi principali che caratterizzano la SLA sono la degenerazione dei motoneuroni delle corna anteriori del midollo spinale, del tronco encefalico e della corteccia cerebrale e la degenerazione del fascio cortico-spinale (piramidale).
La perdita motoneuronale è asimmetrica, esistono aree tipicamente più colpite di altre.
A livello del tronco dell’encefalico sono colpiti tipicamente il nucleo del XII nervo cranico e il nucleo ambiguo (IX, X, XI). Meno frequentemente possono essere interessati i nuclei del V e del VII nervo cranico. Sono generalmente risparmiati i nuclei dei nervi oculomotori (III, IV, VI).
A livello delle corna anteriori del midollo spinale, i più colpiti sono i motoneuroni del gruppo postero-laterale.
Anche il fascio piramidale può essere interessato dalla malattia in modo asimmetrico.
Alla biopsia muscolare, i muscoli colpiti presentano un quadro di atrofia muscolare neurogena con gruppi di fibre atrofiche accanto ad aree con fibre normali e con aumento del tessuto adiposo e connettivale.
Sclerosi Laterale Amiotrofica – Sintomi
I sintomi della SLA sono dovuti alla compromissione sia del primo che del secondo neurone di moto e includono: atrofia muscolare, ipostenia, fascicolazioni e spasticità. La loro distribuzione è variabile all’interno di ogni gruppo muscolare e l’andamento è progressivo. Tipicamente alcune funzioni tendono a essere relativamente risparmiate: i movimenti degli occhi (oculomozione), la sensibilità e il controllo degli sfinteri.
SLA tipica
Rappresenta circa il 50% dei casi. Questa forma ha generalmente un esordio insidioso che può manifestarsi con la riduzione della forza, della destrezza e del trofismo muscolare alle mani: il paziente può avere difficoltà nel manipolare gli oggetti, nello scrivere, nel far girare la chiave nella serratura. Nelle fasi iniziali, può essere colpito soltanto un arto o soltanto un lato del corpo e, in spesso, sono presenti fascicolazioni e crampi muscolari (in qualsiasi distretto, ma soprattutto alle mani). In genere è presente iperreflessia osteotendinea ai quattro arti (segno di compromissione della via piramidale), mentre il segno di Babinski è obiettivabile in circa il 20% dei casi. La sensibilità tattile è conservata. Successivamente i sintomi si estendono in direzione prossimale a interessare tutto l’arto superiore, inclusi i muscoli della spalla, l’arto inferiore, il tronco e i nervi cranici, con comparsa di disfagia e disartria. Circa il 10% dei pazienti sviluppa una demenza di tipo frontale. Il decesso è perlopiù causato dall’instaurarsi di una insufficienza respiratoria di tipo restrittivo e dalle complicanze legate alla disabilità neurologica.
Paralisi Bulbare Progressiva
Nel 25% dei casi circa, la paralisi bulbare progressiva si manifesta come sindrome isolata. I segni bulbari possono tuttavia comparire nel decorso della forma di SLA tipica.
I sintomi legati all’interessamento bulbare della malattia sono: ipotrofia e fascicolazioni della lingua, disartria con difficoltà a pronunciare, in particolare, le consonanti, disfonia fino all’afonia per compromissione delle corde vocali e dei muscoli costrittori della laringe. In fase avanzata è compromessa la motilità linguale e quella del velo palatino. La voce diventa nasale, la deglutizione è compromessa. Più raramente possono occorrere un deficit del VII nervo cranico che si manifesta con riduzione della mimica faciale, in particolare nella zona inferiore del volto, o un deficit del V nervo cranico che determina un deficit dei muscoli masticatori con caduta della mandibola e atrofia dei muscoli massetere e temporale.
Sclerosi Laterale Amiotrofia pseudopolineuropatica o forma agli arti inferiori
Questa forma rappresenta circa il 25-30% dei casi. Si manifesta con un deficit motorio dei muscoli antero-laterali della gamba o talora più prossimale. Il soggetto affetto ha disturbi nella deambulazione con frequenti cadute a terra. Crampi e fascicolazioni possono precedere l’instaurarsi del deficit motorio e dell’atrofia di diversi mesi.
Atrofia Muscolare Progressiva e Sclerosi Laterale Primaria
Alcuni Autori includono nel capitolo della Sclerosi Laterale Amiotrofica anche altre due sindromi: l’atrofia muscolare progressiva e la sclerosi laterale primaria.
L’atrofia muscolare progressiva è un quadro che in passato veniva definito come poliomielite anteriore cronica. L’esordio avviene in età giovanile ed è caratterizzato dalla presenza di fascicolazioni, atrofia agli arti superiori, riduzione dei riflessi osteo-tendinei e assenza di segni piramidali.
La sclerosi laterale primaria è invece un quadro clinico a decorso lentamente progressivo con età di esordio oltre i 50 anni e caratterizzato dalla presenza di spasticità, iper-reflessia osteo-tendinea, segno di Babinski e segni di compromissione bulbare.
Manifestazioni Respiratorie
Quando i muscoli respiratori, in particolare il diaframma, risultano indeboliti dalla malattia, la capacità respiratoria diminuisce. All’inizio è sufficiente la ventilazione a pressione positiva (C-PAP), soltanto di notte o, successivamente, anche di giorno. Nei casi più avanzati è necessario praticare una tracheotomia e ricevere supporto ventilatorio di tipo meccanico.
Manifestazioni neuropsichiatriche
Il 50% dei pazienti affetti da sclerosi laterale amiotrofica manifesta alterazioni di tipo neuro-psicologico conseguenti ad alterazione della corteccia frontale dell’emisfero cerebrale. Le funzioni cognitive più spesso compromesso sono le abilità visuo-spaziali, la capacità di pianificazione, astrazione e problem-solving. Possono manifestarsi anche modifiche della personalità con tendenza alla disinibizione.
Diagnosi
Un esame dirimente è l’elettromiografia, una tecnica neurofisiologica che dimostra e registra l’attività elettrica spontanea o provocata nei muscoli. Essa può evidenziare la presenza di segni di denervazione, sia cronica che in atto, variamente distribuita tra i vari gruppi muscolari. Vengono esaminati i distretti bulbare, cervicale, toracico e lombo-sacrale: è necessaria la documentazione di alterazioni in almeno due di tali distretti e, in ciascun distretto, in almeno due muscoli innervati da diverse radici nervose.
I potenziali evocati motori (PEM) possono documentare la presenza di un’alterazione della via piramidale
L’esame del liquido cerebrospinale è eseguito soprattutto per escludere altre patologie che possono entrare in diagnosi differenziale. In circa la metà dei casi di SLA si osserva un aumento delle proteine.
Anche la risonanza magnetica nucleare è utilizzata per diagnosi differenziale. Non esistono, all’imaging, segni patognomonici che consentano di porre diagnosi di sclerosi laterale amiotrofica. Alle sequenze RM pesate in T2 i tratti corticospinali possono apparire iperintensi: questo aspetto non è però specifico.
La biopsia nervosa o muscolare consente di escludere neuropatie periferiche e miopatie.
La diagnosi di SLA viene posta, secondo i criteri di El Escorial:
- presenza di segni di degenerazione del primo neurone di moto (motoneurone)
- presenza di segni di degenerazione del secondo neurone di moto (motoneurone)
- evoluvità della malattia con documentazione di progressione dei sintomi nello stesso o in altri distretti corporei.
- aver escluso tutte le altre patologie causa di danno al primo e secondo motoneurone.
Sulla base di tali criteri, puramente clinici, si considera:
- SLA sospetta: se esistono segni di compromissione del secondo motoneurone in due o più distretti corporei (i distretti sono: bulbare, cervicale, toracico e lombosacrale).
- SLA possibile: se esistono segni di compromissione del primo o del secondo motoneurone in un solo distretto corporeo oppure segni di compromissione del primo motoneurone in due o più distretti.
- SLA probabile: se esistono segni di compromissione del primo e del secondo motoneurone in almeno due distretti.
- SLA certa: presenza di segni di lesione del primo e del secondo motoneurone sia nel distretto bulbare che in almeno due distretti corporei oppure presenza di segni di primo e secondo motoneurone in almeno tre distretti corporei.
In una revisione del 1998 questi criteri basati esclusivamente su dati clinici sono stati ampliati aggiungendo la definizione di SLA clinicamente probabile con supporto di laboratorio, quando sussistano prove strumentali a favore della diagnosi.
Diagnosi differenziale
- Mieloradiculopatie da spondiloartrosi: si differenziano dalla SLA per la presenza di segni sensitivi e per l’assenza di segni clinici ed eettromiografici di sofferenza di primo e secondo motoneurone in molteplici distretti corporei. Nel caso di spondiloartrosi multiple senza segni sensitivi la diagnosi differenziale può essere difficoltosa: un utile strumento a disposizione del medico è la risonanza magnetica nucleare ma rimane fondamentale una corretta valutazione clinica della storia anamnestica del paziente e l’eventuale evolutività di malattia.
- malattie infettive: da virus dell’immunodeficienza acquisita (HIV), virus della leucemia a cellule T, sifilide, malattia di Lyme.
- altre malattie neurologiche, meno gravi dal punto di vista prognostico, quali sclerosi multipla, sindrome post-poliomielitica, neuropatia assonale motoria acuta, atrofia muscolare spinale.
- polineuropatie di varia eziologia tra cui, in particolare, la neuropatia motoria multifocale, caratterizzata dalla presenza di anticorpi anti GM1 e potenzialmente curabile.
- miopatie: per una corretta diagnosi differenziale è possibile avvalerti di esami ematici e su campioni di urine ed eseguire una biopsia muscolare.
Terapia
L’unico farmaco attualmente autorizzato nel trattamento della sclerosi laterale amiotrofica è il Riluzolo. Esso è un agente anti-glutamaergico (si è già detto che una delle ipotesi di danno coinvolto nell’eziopatogenesi della SLA è legato al danno eccitotossico da glutammato). Studi clinici controllati hanno dimostrato una efficacia nel rallentare l’evoluzione della malattia: tuttavia, il prolungamento della sopravvivenza grazie a questo farmaco è di soli 3 mesi.
La gastrostomia percutanea endoscopica (PEG) prolunga significativamente la sopravvivenza in quanto consente la nutrizione enterale nel paziente disfagico.
Esistono una serie di terapie sintomatiche e palliative che consentono di migliorare la qualità della vita del paziente affetto da sclerosi laterale amiotrofica. Correzione della spasticità, trattamento della scialorrea, antidepressivi per la frequente labilità affettiva, lassativi per la stipsi, controllo del dolore, sostegno respiratorio specie in fase terminale.
Prognosi
Il decorso della sclerosi laterale amiotrofica è del tutto non prevedibile poiché differente da soggette a soggetto. In genere, però, nel giro di pochi anni, si assiste a una perdita progressiva e non reversibile del controllo volontario dei muscoli scheletrici con una paresi ad estensione variabile. Conseguentemente si instaurano: disfagia (perdita della capacità di deglutire), disartria (perdita della capacità di articolare la parola) e insufficienza respiratoria (la paralisi dei muscoli respiratori rende necessario praticare tracheotomie e instaurare ventilazione meccanica).
Va sottolineato che le funzioni sensoriali, il controllo degli sfinteri e le capacità sessuali non vengono intaccate dalla malattia.
Il 75% dei pazienti muore tra i 2 e i 5 anni dalla diagnosi. Il 20% supera i 5 anni ma non arriva ai 20 anni. Soltanto il 5% raggiunge e supera i 20 anni dalla diagnosi.
Le differenze tra SLA e Sclerosi Multipla
La sclerosi laterale amiotrofica e la sclerosi multipla sono due malattie del sistema nervoso centrale a carattere degenerativo ma hanno caratteristiche molto diverse dal punto di vista epidemiologico, fisiopatologico, clinico, di possibilità terapeutiche e prognostico.
- Età: la sclerosi multipla (SM) insorge in genere tra i 20 e i 40 anni con maggiore incidenza nel sesso femminile. La sclerosi laterale amiotrofica insorge invece più tardivamente, tra i 40 e i 70 anni, e più frequentemente il sesso maschile.
- Fisiopatologia: la sclerosi multipla è una patologia demielinizzante, cioè interessa la mielina (il particolare “rivestimento” nervoso che consente una maggiore velocità di conduzione dell’impulso nervoso rispetto alle fibre nervose amieliniche) di qualsiasi struttura del sistema nervoso centrale. La sclerosi laterale amiotrofica, invece, interessa i motoneuroni, centrali e periferici (ovvero le cellule nervose deputate al controllo del movimento). Entrambe la patologie hanno una certa componente autoimmunitaria che si estrinseca, però, su bersagli differenti.
- Caratteristiche cliniche: la sclerosi multipla si manifesta con disturbi neurologici molto variegati, in quanto può essere interessata qualsiasi funzione neurologica (sintomi motori, sensitivi, visivi, disturbi dell’equilibrio, disturbi sfinterici, disturbi sessuali, deficit cognitivi). La sclerosi laterale amiotrofica si manifesta con sintomi prevalentemente motori (atrofia muscolare, deficit di forza, fascicolazioni, spasticità).
- Possibilità terapeutiche: negli attacchi acuti di sclerosi multipla si utilizzano cortisonici mentre l’unico farmaco approvato nel trattamento della SLA è il riluzolo. Le possibilità e le strategie riabilitative sono chiaramente diverse.
- Prognosi: la prognosi della SLA è generalmente molto più severa rispetto a quella della Sclerosi Multipla.
Per approfondire le caratteristiche della Sclerosi Multipla, riferirsi all’articolo del seguente link: Sclerosi Multipla